This is not actually a genomic news, but the new CLARITY brain imaging technique just appeared on Nature is so fascinating that I have to share the video!
This demonstrate how far we have gone in our ability to map the activity of single cells and open amazing possibilities for future studies, promising to provide knowledge on how the brain response to stimuli or coordinate body activities.
The construction of a complete and informative map of the neuron interactions seems feasible and it is a hot topic right now. The US BRAIN project (recently founded with 100 million $ by Obama administration) and the European Human Brain Project (HBP) (founded with 1 billion euros for ten years as one of the EU FET-flegship), are two huge international initiatives, just started to accomplish this ambitious goal.
Someone have already pointed to the brain map as the third revolutionizing achievement after the Human Genome Project and the ENCODE project.
See your brain, with plenty of colourful neuron cell...
This time you can literally say: this is a brilliant idea!
And THIS is amazing science!
Monday, 15 April 2013
Wednesday, 10 April 2013
PubMed Highlight: Updating benchtop sequencing performance comparison
The comparison of available NGS benchtop sequencers continue as every platform rapidly upgrade its performance...or at least claim to have done that!

Accurate evaluation of the present performance of the single technologies is really difficult in a such quickly developing field as NGS...However a snapshot of the NGS technology scenario is of great advantage to everyone to evaluate which one best fit its research needs!
What's new in this paper compared to the last benchmarks from BGI (published on Journal of Biomedicine and Biotechnology) and Wellcome Trust Institute (published on BMC Genomics)?
Two competitors emerge as the leaders in the field: Ion PGM and MiSeq. Data shows data some technological and analytic gaps have been closed, with both platforms performing about the same in term of substitution detection accuracy. MiSeq performance remains better than PGM in detecting small indels (about 100-fold lower error rate), with most of the errors due to troubles in sequencing homopolymers runs. MiSeq still have the lowest cost per Mb, while PGM still the fastest and more flexible.
A good recap of the findings can be found on this post from GenomeWeb, where you can read also the first official answers from both Illumina and Life Tech.

Table from Junemann et al., Nature Biotechnology 31(4): 294-296, April 2013
Tuesday, 9 April 2013
A walking-dead pidgeon and the return of the mummy...you can do NGS starting from any source!!!
Here we are again with some surprising data from NGS studies.
Besides provide some interesting (and, why not, funny) scientific stories, the following news clearly demonstrate how, relying on cut of the edge sample preparation, NGS technology allow sequencing from basically any input material. The ability to obtain a good sequence of an entire genome starting from few nanograms, but even picograms, of input DNA open the way to amazing application. From cancer genomics, to prenatal screening, the ability of sequence very little amount of cell-free DNA that is present in circulating blood as already provide really exciting results. This kind of techniques promise great improvements also in epidemic controls and contamination/pathogens detection in foods, water or any other samples.
The topic of sequencing from nanograms or picograms is extensively covered in this post on CoreGenomics, that provides also some examples of recently published papers and dicuss about the new library preparation kits that make NGS sequencing from low input DNA fast and easy!
Now the stories!
 We have reported on September 2012 about Revive and Restore (see the our post here), a company founded with the ambitious and controversial aim of sequence and reconstruct the genome of extinct species with the ultimate goal of eventually bring them back to life.
We have reported on September 2012 about Revive and Restore (see the our post here), a company founded with the ambitious and controversial aim of sequence and reconstruct the genome of extinct species with the ultimate goal of eventually bring them back to life.
We were not sure how far this initiative would have gone, but the last month they surprised us again with the announcement of an actual project to resurrect the extinct passenger pidgeon.
The topic of sequencing from nanograms or picograms is extensively covered in this post on CoreGenomics, that provides also some examples of recently published papers and dicuss about the new library preparation kits that make NGS sequencing from low input DNA fast and easy!
Now the stories!
 We have reported on September 2012 about Revive and Restore (see the our post here), a company founded with the ambitious and controversial aim of sequence and reconstruct the genome of extinct species with the ultimate goal of eventually bring them back to life.
We have reported on September 2012 about Revive and Restore (see the our post here), a company founded with the ambitious and controversial aim of sequence and reconstruct the genome of extinct species with the ultimate goal of eventually bring them back to life.We were not sure how far this initiative would have gone, but the last month they surprised us again with the announcement of an actual project to resurrect the extinct passenger pidgeon.
Ben Novak, a 26-year-old genetics, has received support from the company to achieve the goal of sequencing the entire DNA of this species from a tissue sample received from the Chicago's Field Museum in 2011. Working with evolutionary biologist Beth Shapiro at the University of California, Santa Cruz, they plan to complete the genome of passenger pidgeon and its closest living relative, the band-tailed pidgeon. The extinct DNA will then be aligned to the living one to identify all the differences and finally a massive mutagenesis will be performed on band-tailed pidgeon DNA to re-create the complete sequence of the passenger species.
However the idea not only require hard work, but it could really get dicey. Indeed, according to Shapiro, "because the last common ancestor of the two species flew about 30 million years ago, their genomes will likely differ at millions of locations." Fitting the pieces together will be grueling, if not impossible. GenomeWeb have a post on this and Wired also has covered the story in this article from Kelly Servick.

However one consider this real science or fantasy science, the general topic of de-extinction is getting increasing attention now that the DNA sequencing technology allow to effectively assemble genomes from ancient and degraded samples (remember for example the Neanderthal genome or the Mammoth genome). The collection of DNA from different species is a part of some huge projects intended to preserve and study biodiversity and experts are now discussing if and eventually how we have to deal with species that go extinct over time. If we as human race are responsible for the disappearing of a specific organism and we have the ability to bring it back to life, should we do this? What are the risks of re-introduce extinct species in our ecosystem?
 Recently a TEDx event has been organized exploring the topic of de-extinction. and the recent advances in the field have received attention also from the National Geographic and The New York Times (this one dealing with bring an extinct frog back to life). Revive and Restore has a list of candidate organisms waiting for de-exctinction and is searching for collaborators! Looking at their list I may like to see a Dodo walking again in the garden...but want to raise my doubts about a tooth-saber cat!!
Recently a TEDx event has been organized exploring the topic of de-extinction. and the recent advances in the field have received attention also from the National Geographic and The New York Times (this one dealing with bring an extinct frog back to life). Revive and Restore has a list of candidate organisms waiting for de-exctinction and is searching for collaborators! Looking at their list I may like to see a Dodo walking again in the garden...but want to raise my doubts about a tooth-saber cat!!
 The second news is directly from scientific literature. In their paper recently published on Journal of Applied Genetics, Khairat et al. from the University of Tubingen, report the first metagenome analysis on ancient egyptian mummies. Their dataset comprise seven sequencing experiments performed on DNA obtained from five randomly selected Third Intermediate to Graeco-Roman Egyptian mummies (806 BC-124AD) and two unearthed pre-contact Bolivian lowland skeletons. Analyzing the data their were able to identify different genetic materials from bacteria, presumibly due to contamination from mummies conservation procedures, and also from plants, potentially associated with their use in embalming reagents. The paper demonstrates that also DNA from ancient mummies, could be a proper template for NGS sequencing, despite its age and the several treatment performed on the samples in the course of the conservation protocols.
The second news is directly from scientific literature. In their paper recently published on Journal of Applied Genetics, Khairat et al. from the University of Tubingen, report the first metagenome analysis on ancient egyptian mummies. Their dataset comprise seven sequencing experiments performed on DNA obtained from five randomly selected Third Intermediate to Graeco-Roman Egyptian mummies (806 BC-124AD) and two unearthed pre-contact Bolivian lowland skeletons. Analyzing the data their were able to identify different genetic materials from bacteria, presumibly due to contamination from mummies conservation procedures, and also from plants, potentially associated with their use in embalming reagents. The paper demonstrates that also DNA from ancient mummies, could be a proper template for NGS sequencing, despite its age and the several treatment performed on the samples in the course of the conservation protocols.
However the idea not only require hard work, but it could really get dicey. Indeed, according to Shapiro, "because the last common ancestor of the two species flew about 30 million years ago, their genomes will likely differ at millions of locations." Fitting the pieces together will be grueling, if not impossible. GenomeWeb have a post on this and Wired also has covered the story in this article from Kelly Servick.

However one consider this real science or fantasy science, the general topic of de-extinction is getting increasing attention now that the DNA sequencing technology allow to effectively assemble genomes from ancient and degraded samples (remember for example the Neanderthal genome or the Mammoth genome). The collection of DNA from different species is a part of some huge projects intended to preserve and study biodiversity and experts are now discussing if and eventually how we have to deal with species that go extinct over time. If we as human race are responsible for the disappearing of a specific organism and we have the ability to bring it back to life, should we do this? What are the risks of re-introduce extinct species in our ecosystem?
 Recently a TEDx event has been organized exploring the topic of de-extinction. and the recent advances in the field have received attention also from the National Geographic and The New York Times (this one dealing with bring an extinct frog back to life). Revive and Restore has a list of candidate organisms waiting for de-exctinction and is searching for collaborators! Looking at their list I may like to see a Dodo walking again in the garden...but want to raise my doubts about a tooth-saber cat!!
Recently a TEDx event has been organized exploring the topic of de-extinction. and the recent advances in the field have received attention also from the National Geographic and The New York Times (this one dealing with bring an extinct frog back to life). Revive and Restore has a list of candidate organisms waiting for de-exctinction and is searching for collaborators! Looking at their list I may like to see a Dodo walking again in the garden...but want to raise my doubts about a tooth-saber cat!! The second news is directly from scientific literature. In their paper recently published on Journal of Applied Genetics, Khairat et al. from the University of Tubingen, report the first metagenome analysis on ancient egyptian mummies. Their dataset comprise seven sequencing experiments performed on DNA obtained from five randomly selected Third Intermediate to Graeco-Roman Egyptian mummies (806 BC-124AD) and two unearthed pre-contact Bolivian lowland skeletons. Analyzing the data their were able to identify different genetic materials from bacteria, presumibly due to contamination from mummies conservation procedures, and also from plants, potentially associated with their use in embalming reagents. The paper demonstrates that also DNA from ancient mummies, could be a proper template for NGS sequencing, despite its age and the several treatment performed on the samples in the course of the conservation protocols.
The second news is directly from scientific literature. In their paper recently published on Journal of Applied Genetics, Khairat et al. from the University of Tubingen, report the first metagenome analysis on ancient egyptian mummies. Their dataset comprise seven sequencing experiments performed on DNA obtained from five randomly selected Third Intermediate to Graeco-Roman Egyptian mummies (806 BC-124AD) and two unearthed pre-contact Bolivian lowland skeletons. Analyzing the data their were able to identify different genetic materials from bacteria, presumibly due to contamination from mummies conservation procedures, and also from plants, potentially associated with their use in embalming reagents. The paper demonstrates that also DNA from ancient mummies, could be a proper template for NGS sequencing, despite its age and the several treatment performed on the samples in the course of the conservation protocols.All start from high quality reads in NGS!
If you are performing an NGS based experiment, first of all you want to be sure that your are starting from high quality raw data. Current technology have achieved outstanding robustness but the quality check on sequencing reads remain the first step in every analysis.


Several tools are available that return stats and graphs from analysis of your fastq files and, inspired by a new paper just appeared on PLoS ONE, I just report want to report a couple of solutions that I found useful.
First is FastQC. This is a relative simple tools which take your fastq or bam/sam file and report all the essential stats you need to be sure that nothing has gone wrong with your sequencing. It's based on Java and so it can easily run on almost every platform without the need for tricky installation steps.
You can find this from the official web page at Babraham Bioinformatic Institute.

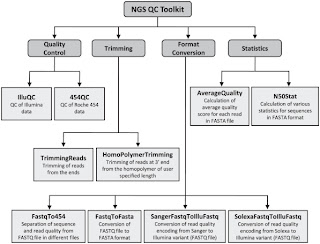
Second is NGS QC Toolkit. This is a set of tools for the quality control of next generation sequencing data. It accept data in the popular fastq format and provide with detailed results in the form of tables and graphs. Moreover it allows filtering of high-quality sequence data and includes few other tools, which are helpful in NGS data quality control and analysis (format conversion and trimming of the reads for example).
It is developed by the Indian National Institute of Plant Genome Research and you can find it at its official page here.
Also take a look at the official paper published on PLoS ONE in 2012 by Patel RK & Jain M

Third is this recent QC-chain tool that have cited above. The tool comprise a set of user-friendly tools for quality assessment and trimming of raw reads (Parallel-QC). Moreover it has an interesting feature that allows identification, quantification and filtration of unknown contamination to get high-quality clean reads. Authors stated that the tool was optimized based on parallel computation, promising that processing speed is significantly higher than other QC methods...This could be really useful if you routinely deal with a huge volume of data.
QC-chain is developed by the Computation Biology Team at Qingdao Institute of Bioenergy and Bioprocess Technology, and can be found here at the official web page.
This one also have an official paper published on PLoS ONE in 2013 by Zhou Q et al.

Subscribe to:
Posts (Atom)